摘要
间接免疫荧光法是检测抗核抗体的参考方法,然而检测过程标准化程度低、检测结果报告一致性差是临床实验室面临的主要挑战。随着抗核抗体荧光模型国际共识组织对各荧光模型进行标准化命名,以人喉癌上皮细胞为基质的间接免疫荧光法实验由手工操作逐步转向全自动仪器操作以及计算机辅助诊断系统的临床应用,实验室对该项目的规范化检测及结果报告提出了新的要求。本共识从检验项目开展前、检验过程和结果报告3个方面进行阐述并提出相应意见,旨在进一步推进该项目在我国实验室检测与报告的规范化与标准化。
抗核抗体(antinuclear antibody,ANA),广义上是指针对真核细胞各种抗原成分的自身抗体,是系统性红斑狼疮(systemic lupus erythematosus,SLE)、干燥综合征(Sjögren′s syndrome,SS)、系统性硬化症(systemic sclerosis,SSc)、混合性结缔组织病(mixed connective tissue disease,MCTD)和特发性炎症性肌病(idiopathic inflammatory myopathy,IIM)等自身免疫病的重要血清学标志物。这些疾病由于与ANA的密切关系,也被称为抗核抗体相关风湿病(ANA-associated rheumatic diseases,AARD)。
ANA的检测对于AARD的诊断和疾病分类具有重要意义。2019年欧洲抗风湿病联盟/美国风湿病学会(European League Against Rheumatism/American College of Rheumatology,EULAR/ACR)SLE 分类标准的准入条件是:以人喉癌上皮细胞(human epithelioma-2,HEp-2)为基质的间接免疫荧光法(immunofluorescence assay,IFA)检测ANA阳性(滴度≥1∶80),或与HEp-2 IFA具有等效敏感性的固相ANA筛查试验阳性[1 ]。HEp-2细胞是包含100多种自身抗体靶抗原的“天然阵列”,以其作为基质对ANA进行IFA检测,敏感性高,是目前公认的ANA检测的参考方法[2 ]。
作为自身抗体检测实验室广泛开展的检测项目,HEp-2 IFA检测ANA仍存在手工操作占比重高、检测标准化程度低、实验室间检测结果差异大和需要实验室配备专业读片人员等问题。近年来,在促进HEp-2 IFA检测ANA的规范化和标准化上,国际和国内专家学者以及相关企业作出巨大的努力。ANA荧光模型国际共识组织(the International Consensus on ANA Patterns,ICAP)提出了以AC(anti-cell)编号的ANA荧光模型的命名[3 ]、临床相关性[4 ]以及结果规范化报告指南[5 ],欧洲检验医学联合会、欧洲自身免疫标准化促进会和ICAP共同发表了对ANA检测的推荐意见[6 ];我国参加ANA室间质量评价的实验室数量与合格率不断提升[7 , 8 ];全自动荧光制片机以及计算机辅助诊断(computer-assisted diagnosis,CAD)系统在临床工作中的普及率逐步增高[9 , 10 ]。然而,目前我国仍缺乏HEp-2 IFA实验室规范化检测ANA的指南。
在临床实验室逐步由HEp-2 IFA检测ANA的手工操作发展为自动化仪器检测结合CAD系统辅助荧光读片的现状下,中国中西医结合学会检验医学专业委员会和上海市医学会检验医学专科分会(以下简称“专委会”)共同制定了《间接免疫荧光法用于抗核抗体实验室检测的中国专家共识(2023年)》(以下简称《共识》),旨在进一步提高HEp-2 IFA检测ANA的质量和结果的一致性,为各级医院从事自身抗体检测相关工作的医务工作者和科研人员提供切实有效的建议。
《共识》指定过程、证据概述和专家评分
一、制定过程
《共识》制定过程分4步进行:首先专委会专家讨论确立了本共识主要内容,即从检验项目开展前(性能验证、临床沟通、人员培训、仪器校准和标准化操作程序制定)、检验过程(实验室检测、质量评价、批号验证)和结果报告(荧光模型报告、滴度特异性似然比、报告模板)3个方面进行论述;结合中国合格评定国家认可委员会(China National Accreditation Service for Conformity Assessment,CNAS)对医学实验室ISO15189评审要求和文献学习以及多家医疗机构的临床工作实践形成草案,后经专委会讨论并修改形成推荐意见分级的评估、制定及评价(grading of recommendations assessment,development and evaluation,GRADE)方法的证据质量分级[11 ];草案提交专家组评分后统计每条建议的专家认可度;最终对所有问题进行公开讨论并修改完善后定稿。
二、证据级别评价标准
共识中参考文献证据按照GRADE系统共分3级[11 ],表述如下。
证据质量高(A):证据基于多项随机临床试验或荟萃分析,进一步的研究不太可能改变对效果估计的信心。
证据质量中(B):证据基于单项随机临床试验或多项非随机对照研究,进一步的研究可能会对效果估计的信心产生重要影响并可能改变评估结果。
证据质量低或非常低(C):仅为专家意见,或基于注册研究结果,进一步的研究很有可能影响对效果估计的可信度,且很可能改变评估结果。
三、专家评分
专家对于共识意见( 表1 )的认可度以Delphi评分进行打分(分值0~10分),0分表示完全不赞同,10分表示完全赞同(0~2分反对,3~4分不推荐,5~6分酌情推荐,7~8分推荐,9~10分强烈推荐)。专家认可度以所有专家打分的平均数±标准差(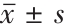 )及其95%可信区间(confidence interval,CI)表示。
)及其95%可信区间(confidence interval,CI)表示。

检验项目开展前
一、性能验证
HEp-2 IFA是ANA检测的筛查试验,属于临床免疫学定性检测项目,其结果通常包含荧光模型和半定量滴度。影响HEp-2 IFA检测ANA结果的因素较多,如:商品化试剂盒中基质HEp-2细胞,在培养、固定和透化过程中导致的细胞抗原反应性不同[12 , 13 ];荧光二抗的浓度、与人免疫球蛋白G(immunoglobulin G,IgG)结合的特异性、荧光素/蛋白摩尔比不同[14 ];洗液、封片介质不同;实验操作及环境温度影响;显微镜光源不同(发光二极管灯、汞灯或金属卤化物灯)或是否采用CAD系统等。因此,临床实验室在项目开展前,需要对产品进行性能验证,验证内容至少包括:符合率、精密度和参考区间[15 , 16 ]。使用CAD系统以及全自动荧光制片机的实验室也需要在项目开展前完成相关比对和验证工作。
(一)符合率
HEp-2 IFA检测ANA的符合率通常选择同品牌试剂进行比对,优先选择已开展该项目且通过CNAS认可的医学实验室。比对样本要求[15 , 17 ]:(1)至少30份血清样本,其中阴性、弱阳性以及阳性样本各10份;(2)阴性样本应包含显微镜下有荧光,且强度不强于阴性质控和荧光模型不清的样本;(3)阳性(含弱阳性)样本建议尽量覆盖临床常见或有重要临床意义的荧光模型[6 ],且涵盖细胞核荧光模型以及细胞浆荧光模型,包括但不限于:均质型(AC-1)、致密细颗粒型(AC-2)、着丝点型(AC-3)、核颗粒型(AC-4,5)、核点型(AC-6,7)、核仁型(AC-8,9,10)、核膜型(AC-11,12)、胞浆线性/肌动蛋白型(AC-15)、胞浆颗粒型(AC-19,20)、胞浆网状/线粒体样型(AC-21);(4)阳性样本建议尽量包含临床常见特异性抗体阳性样本,如:抗双链DNA(double strand deoxyribonucleic acid,dsDNA)抗体、抗干燥综合征抗原A(Sjögren′s syndrome related antigen A/Ro60,SS-A/Ro60)抗体、抗斯密斯(Smith,Sm)抗体、抗U1小核核糖核蛋白(U1 small nuclear ribonucleoprotein,U1-snRNP)抗体、抗着丝粒蛋白-B(centromere proteins-B,CENP-B)抗体、抗可溶性酸性磷酸化核蛋白100(speckled protein 100,Sp100)抗体、抗跨膜糖蛋白210(glycoprotein-210,gp210)抗体、抗核糖体P蛋白(ribosomal P,Rib-P)抗体、抗组氨酰tRNA合成酶(histidyl tRNA synthetase,Jo-1)抗体和抗线粒体抗体-M2型(anti-mitochondrial antibody-M2,AMA-M2)。若在某一时段内,所收集样本暂时无法满足上述要求,可用室间质评样本或血清基质质控品替代,但仍需在半年内完成临床样本收集和符合率验证[6 , 15]。
将比对样本随机分成5组,每天检测1组,每组6个样本,连续5 d,按照患者样本检测程序进行检测,阴阳性一致,阳性样本荧光模型相同且在上下一个滴度范围内为符合,计算Kappa值和符合率[15 , 18 ],Kappa值≥0.8且符合率≥80%为可接受标准。
(二)精密度
HEp-2 IFA检测ANA结果中滴度是以量值形式表达的定性结果,需进行精密度验证,包括重复性和中间精密度。应评估阴性、弱阳性和阳性3个水平,阳性(含弱阳性)样本可选用临床常见荧光模型,如:均质型或核颗粒型。重复性和中间精密度可同时验证,每天检测1个分析批,每个样本每天重复检测3~5次,连续检测5 d[19 ]。当结果为二分类变量(阴/阳性)或有序分类变量(滴度)时,精密度验证所计算的是与原始结果的符合率,即阴阳性符合,阳性样本荧光模型一致且上下一个滴度范围内的比率。
精密度验证的可接受标准为所用厂商检验方法声明的标准,若无可用的厂商标准时,实验室可根据临床诊疗的质量要求确定可接受标准,建议可接受标准为与原始结果一致率≥80%(建议仪器制片法一致率≥90%)。
(三)参考区间
参考区间验证可通过检测至少20份健康人的血清样本,验证试剂盒厂家提供的参考区间,20例测定值中落在参考区间外的测定值不超过2个,参考区间可直接使用[20 ]。实验室也可设置适合本地区的参考区间,建议使用非参数的百分位数方法建立临界值,推荐采用95的百分位数[21 ],大于临界值判断为阳性。
(四)CAD系统验证
采用CAD系统进行HEp-2 IFA检测ANA结果报告须对该系统进行性能验证[6 ]。
1.阴阳性判读:需进行自动化判读系统与人工显微镜下判读阴阳性符合率的评估。实验室可自行设立判读符合率要求,但至少应≥80%[9 , 22 , 23 ]。
2. 荧光模型判读:以读片人员判读结果进行报告,不推荐直接采用CAD荧光模型判读结果[24 ]。
3. 滴度判读:CAD系统荧光强度与样本中自身抗体滴度具有相关性。ANA阳性样本CAD估算滴度与终点滴度一致性75.9%~97.8%[9 ,- 10 , 25 ],但估算滴度通常高于终点滴度1~2个稀释度[26 ]。医学实验室若直接使用CAD系统的估算滴度进行报告,则需要在项目开展前对其与终点滴度进行符合率验证。
若符合率未达到要求,实验室可依据荧光强度和终点滴度的对应关系,建立合适的区间,并用独立样本进行符合率评估。在此过程中值得注意的是:(1)不同荧光模型中相同终点滴度样本的荧光强度分布差异较大,因此在实验室应至少建立并验证均质型、核颗粒型、着丝点型和核仁型4种常见荧光模型的估算滴度符合率[9 , 26 , 27 ]。(2)通过PASS 15计算样本量,构建受试者工作特征曲线下面积(area under curve,AUC)的双侧95%可信区间,根据既往研究可知AUC约为0.96[9 ],用Clopper Pearson精确概率法构建可信区间,当可信区间宽度为0.1时,每组所需阳性份数为30份。例如:倍比稀释系统下构建核颗粒型荧光强度与滴度对应值时,至少收集终点滴度为1∶80、1∶160、1∶320、1∶640、1∶1 280和>1∶1 280共6组样本,每组各30份作为训练集。(3)验证样本量可按照与训练集比例1∶2、3∶7或者1∶4进行设定。
(五)人员比对
1. 实验操作比对:当实验室采用手工法进行HEp-2 IFA检测ANA操作时,应在检验人员完成相关实验原理、操作步骤等培训后,进行不同人员间操作比对。比对样本应包括阴性、弱阳性和阳性样本各至少3份。比对的阳性样本中应包含低滴度抗SS-A/Ro60抗体阳性核颗粒型血清[6 , 28 ],以及胞浆型荧光模型血清。若实验室采用全自动荧光制片机,也应进行仪器与实验操作人员间的比对。
2. 结果判读比对:HEp-2 IFA检测ANA的结果判读具有一定主观性,因此建议采用双人在显微镜下或CAD屏幕上独立阅片,结果不一致时由两人讨论后进行结果报告。在项目开展前可利用性能验证中符合率验证的荧光片,进行不同人员判读结果比对。人员间阴阳性以及荧光模型判读符合率应≥80%。
当项目开展后,仍需定期进行人员的培训考核和人员间制片操作和结果判读的比对。
(六)其他验证及比对
在项目开展前除了对上述方面进行验证和比对外,还应注意下述情况。
1. 血清和血浆一致性比对:ANA检测推荐样本类型为血清[29 ],若试剂说明书中声明可检测血浆中ANA,且实验室ANA检测样本类型为血清或血浆时,需要验证血浆与血清结果一致性。可收集ANA阴性、弱阳性和阳性样本(同一患者收集配对血清与血浆样本),每组各10份,一致性100%则验证通过[30 ]。另外,对不同抗凝剂血浆与血清的一致性,应分别进行验证。
2. 血清干扰因素验证:溶血、脂血、黄疸、微生物污染或有可见微粒的血清,不建议用于HEp-2 IFA检测ANA,若试剂说明书中声明干扰因素不影响实验,且实验室允许采用含相关干扰物质血清进行样本检测,则需对干扰物质的干扰能力进行验证。可收集5份样本,包括ANA阴性、弱阳性和阳性,与ANA阴性且含有高浓度待验证干扰物的血清混合(干扰物加入量<10%),对照组加入等量ANA阴性健康人血清。加入干扰物阳性组结果与阳性对照组之间符合率≥80%,加入干扰物阴性组与阴性对照组结果均为阴性,则该干扰物不影响实验[30 ]。
3. 荧光片保存条件:HEp-2 IFA检测ANA荧光片的荧光强度及基质细胞形态对结果判断至关重要,若不能及时读片或者临床实验室拟保存荧光片以备复核,需要对荧光片的保存时间和温度条件进行评估。荧光片通常需要2~8 ℃避光保存,在保存条件和时间内,必须阴阳性符合,且阳性样本滴度不下降。
(七)其他需要进行性能验证的情况
除了项目开展前需要进行性能验证外,在项目正式开展后,如出现严重影响检测程序的情况,包括但不限于:仪器主要部件故障、仪器搬迁或更新、试剂升级、环境严重失控,纯水系统改变等,应进行性能评估,至少包括符合率和精密度。
二、结果报告、解读及临床沟通
临床实验室在开展HEp-2 IFA检测ANA前,应做好充分的临床沟通,除了检测样本类型、医嘱申请、检测频率、报告周转时间等基本情况外,还需包含以下内容。
(一)报告荧光模型及临床意义
HEp-2 IFA作为ANA筛查实验,其荧光模型有助于指导下一步特异性血清学分析[21 ]。例如:胞浆网状/线粒体样型(AC-21),可建议临床检测AMA-M2抗体;核多点型(AC-6),若临床怀疑原发性胆汁性胆管炎(primary biliary cirrhosis,PBC),可进一步检测抗Sp100抗体和抗早幼粒细胞白血病蛋白(promyelocytic leukemia,PML)抗体,若怀疑皮肌炎,建议检测抗核基质蛋白2(nuclear matrix protein 2,NXP-2)抗体。ICAP中文网站(www.anapatterns.cn)列有不同荧光模型以及相关靶抗原的临床相关性,可供实验室以及临床医生查阅。
部分荧光模型的临床意义较为明确,但其对应的抗体不在实验室常规检测的项目中,实验室应与临床医生沟通上述荧光模型的临床相关性。如:致密细颗粒型(AC-2)常见靶抗原为致密细颗粒70(dense fine speckles 70,DFS70),该抗体单阳性少见于系统性结缔组织病患者;核有丝分裂器蛋白(nuclear mitotic apparatus,NuMA)样型(AC-26)靶抗原主要为NuMA1,常见于SS和SLE患者,也可见于未分化结缔组织病、局限性皮肤型SSc和类风湿关节炎(rheumatoid arthritis,RA)等患者;棒环状型(AC-23)靶抗原主要为肌苷单磷酸脱氢酶 2 型(inosine monophosphate dehydrogenase type 2,IMPDH2),可在接受聚乙二醇干扰素-α/利巴韦林联合治疗的丙型肝炎病毒感染患者中检出。
(二)滴度报告
1. 滴度稀释系统:不同试剂可能采用不同稀释体系,如:倍比稀释系统(1∶40、1∶80、1∶160、1∶320、1∶640、1∶1 280等)或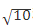 稀释系统(1∶100、1∶320、1∶1 000、1∶3 200、1∶10 000等),不同稀释系统的结果间不能直接比较。建议开展多中心、大样本HEp-2 IFA不同滴度系统研究,了解不同滴度系统强阳性、中等阳性、弱阳性滴度范围及表观健康人情况。
稀释系统(1∶100、1∶320、1∶1 000、1∶3 200、1∶10 000等),不同稀释系统的结果间不能直接比较。建议开展多中心、大样本HEp-2 IFA不同滴度系统研究,了解不同滴度系统强阳性、中等阳性、弱阳性滴度范围及表观健康人情况。
2. 起始筛查稀释度:倍比稀释系统中起始筛查稀释度1∶160区分正常人与结缔组织病患者特异性较高[21 , 31 , 32 , 33 ];1∶80作为筛查稀释度敏感性高,也是2019年EULAR/ACR SLE 分类标准的准入条件[1 ]。稀释系统常以1∶100作为起始筛查稀释度,正常人群在该稀释系统下1∶320稀释度时阳性率<5%[34 ]。儿童和成人SLE由于在同一起始筛查稀释度下检测ANA阳性率差异无统计学意义,其起始筛查稀释度可与成人相同[6 , 35 , 36 , 37 ],但需与临床沟通确认。
3. 最大稀释度:ANA滴度不适用于判断疾病活动度和治疗反应性[21 , 29 , 38 ],但高滴度ANA可增加HEp-2 IFA诊断特异性,因此实验室报告的最大稀释度至少应≥1∶1 280(倍比稀释系统)[21 , 29 ]或≥1∶3 200(稀释系统)[5 , 34 , 39 ]。
(三)正常人群HEp-2 IFA检测ANA阳性率分布特点
正常人群中,HEp-2 IFA滴度≥1∶40、≥1∶80、≥1∶160和≥1∶320的ANA阳性率分别为20%~30%、10%~12%、5%和3%[40 ]。我国体检人群中HEp-2 IFA检测ANA滴度≥1∶80或≥1∶100时,ANA阳性率是7.4%~13.5%[41 , 42 , 43 , 44 , 45 , 46 ]。ANA阳性率与年龄呈正相关,50岁以上人群阳性率明显高于50岁以下人群,且女性高于男性。我国正常人群不同年龄段ANA≥1∶80或≥1∶100时的阳性率:<10岁儿童2.2%~5.5%(男1.6%~2.9%;女2.8%~8.6%),10~50岁5.0%~11.1%(男2.7%~7.9%;女7.5%~14.8%),>50岁11.3%~19.1%(男6.2%~14.0%;女16.3%~25.3%)[42 , 44 , 45 ]。
(四)HEp-2 IFA检测ANA与疾病活动度
HEp-2 IFA检测ANA的结果,包括荧光模型和滴度,均可能会随着SLE患者疾病活动性而改变。有研究显示,高滴度ANA荧光和均质型(AC-1)荧光模型与疾病活动有关,低滴度和细颗粒型(AC-4)和疾病缓解相关[47 ],但HEp-2 IFA检测ANA的结果与疾病活动度的关系仍需更多证据来明确,目前该检测仍应以辅助诊断为目的,而非作为疾病活动度评估或治疗反应性监测[48 , 49 ]。
三、人员培训和仪器校准
(一)人员培训
从事HEp-2 IFA检测ANA实验操作以及结果判读的人员应参与培训并通过考核,获得相关授权后,方可独立工作[50 ]。人员培训的内容应包括但不限于:(1)实验基本原理和操作步骤;(2)仪器和显微镜的操作、维护保养和处理一般性故障;(3)质量控制及失控处理;(4)荧光模型判读及相关靶抗原临床意义;(5)试剂更换批号比对。
(二)仪器校准
HEp-2 IFA检测ANA手工或仪器操作时,应注重对移液器或加样针的校准。校准时需涵盖整个实验过程所涉及的加样体积的加样准确性和精密度,并使其在制造商声明的范围之内。仪器法进行HEp-2 IFA法检测ANA时,还需进行携带污染率评估。显微镜以及CAD系统通常采用荧光显微镜校准玻片或光功率计对光强度进行测量和校准,校准后光强度应在仪器说明书所规定的光强度范围内,偏差≤5%。所有仪器的校准频率至少每年1次,对于仪器使用频繁的实验室,可增加校准频率[50 ]。
四、建立《标准化操作程序》
在HEp-2 IFA检测ANA检验程序常规应用前,应建立《标准化操作程序》(standard operation procedure,SOP)以指导整个检验过程。SOP文件内容包括但不限于:(1)项目信息:包括项目名称、检测方法及基本原理、试剂、荧光显微镜、自动化仪器(如使用自动荧光制片机、CAD系统)等。(2)样本要求:检测样本类型及储存要求。(3)室内质控程序及可接受标准。(4)实验操作步骤:包括具体实验步骤和注意事项等。(5)项目性能参数:包括参考区间和干扰物等。(6)结果报告:包括结果解释、临床意义、备注信息等。(7)其他信息:如荧光片保存条件等。
如使用自动化仪器(自动化荧光制片机、CAD系统),还应建立仪器SOP,内容包括但不限于仪器校准要求、仪器操作步骤及维护保养、常见故障及处理措施。
检验过程
一、实验室检测
实验室HEp-2 IFA检测ANA的标准化和规范化是检测结果准确的重要保障,在实验过程中应注意以下事项。
(一)实验前
(1)确认环境温湿度符合检测体系的要求,通常室温18~26 ℃,湿度30%~50%;(2)测试样本和试剂应平衡至室温,荧光片进行室温平衡时不能从包装的铝膜中拆出;(3)按试剂说明书配制洗液,洗液应充分混合;(4)可用pH试纸或pH计,确认洗液的pH值为7.2~7.4。
(二)实验中
(1)每批实验必须有血清基质的质控品;(2)加样时防止加样枪接触到细胞基质,避免产生气泡,液体应覆盖整个孔,且不能外溢到相邻孔中;(3)洗片时,注意不能将洗液直接对着孔内冲洗,避免损伤基质抗原;(4)加荧光二抗前,应尽量吸干玻片上多余的洗液,避免影响二抗浓度;(5)注意观察玻片的疏水性,保持荧光片上的试剂呈液滴状态,防止液体散开并外流至其他孔中;(6)实验过程中禁止用手指触碰荧光片的细胞基质;(7)封片时,避免孔内有气泡产生,影响读片;(8)完成实验后,应立刻读片,如不能立即读片,应按荧光片保存条件的规定进行保存;(9)实验过程由全自动仪器完成时,实验过程中应观察每个样本孔的样本及试剂是否加样准确,保证实验过程洗液充足,没有干片现象。
(三)结果判读
(1)光源为汞灯的显微镜接通电源后须预热10~15 min,汞灯电源开、关时间应间隔30 min以上;(2)注意显微镜的维护保养,汞灯显微镜应记录每次使用时长,到达使用寿命及时更换;(3)必须在质控结果符合要求的情况下,进行样本结果判读;(4)低倍镜(20倍物镜)判读阴阳性结果,高倍镜(40倍物镜)判读荧光模型;(5)采用CAD系统实验室,CAD判读结果应由读片人员复核后进行报告;(6)结果判读建议“双人双核”,即双人独立阅片后相互核对,结果不一致时由两人讨论后进行结果报告;(7)若手工记录判读结果再录入电脑,需要双人核对,确保结果录入的准确性。
(四)前带效应
HEp-2 IFA检测ANA偶见前带效应,即由于血清中自身抗体浓度过高,导致荧光呈阴性或较弱染色[29 , 51 ]。同时进行至少2个稀释度检测时,若出现较高稀释度荧光强度高于低稀释度荧光强度,则提示存在前带效应,可进一步进行梯度稀释,有效避免前带效应对结果的影响[52 ]。另外,如图1所示荧光染色间期细胞的细胞核核周染色较亮中间暗,或荧光基质片显微镜下中央区域暗、边缘区亮,提示可能存在前带效应,可进一步稀释确认[53 ]。
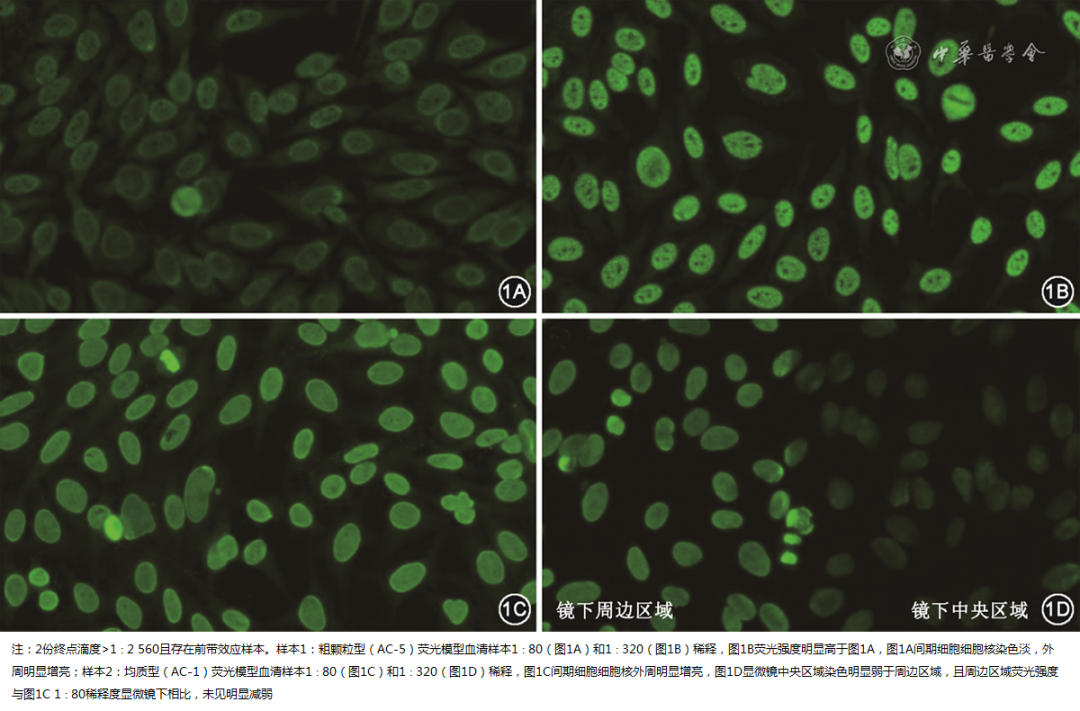
图1临床常见前带效应荧光表现(间接免疫荧光染色×200)
(五)试剂和洗液储存条件
荧光片和试剂需2~8 ℃保存,试剂盒在冰箱内应避免贴壁造成温度过低。荧光二抗避光保存。稀释后的洗液也可2~8 ℃保存,保存时间可参考试剂说明书,通常不超过1周,若洗液出现浑浊或沉淀则不可使用。
二、质量控制
(一)室内质控
1. 室内质控品:HEp-2 IFA检测ANA实验的室内质控品要求血清基质,每批次实验推荐做3个水平,即阴性、弱阳性(倍比稀释系统:1∶80或1∶160,稀释系统:1∶100或1∶320)和阳性(倍比稀释系统:≥1∶640,稀释系统:≥1∶3 200);如条件有限,则至少做2个水平的质控,即阴性和弱阳性[13 ]。阳性质控品荧光模型可以为临床常见的单一荧光模型,如核颗粒型或均质型。实验室可使用商品化的质控血清,或自制混合血清作为室内质控。不推荐仅采用商品化试剂盒中自带的对照品作为实验检测的室内质控品,其存在以下不足:(1)试剂盒中的对照品通常只包含阳性和阴性2种,无弱阳性水平;(2)无须进行稀释,故无法监测实验过程中血清的稀释步骤;(3)阴阳性对照随试剂批号更换,无法保证一致性[6 ]。
2. 自制室内质控品:可采用HEp-2 IFA检测ANA阴性、弱阳性和阳性混合血清或来源于单一患者的血清作为室内质控品[15 , 54 ],充分混匀分装后对自制质控品进行均匀性和稳定性验证。(1)均匀性验证:即瓶间差分析,随机抽取10份分装质控品,每个样本重复检测2次,比较20个结果的符合率,要求符合率100%;(2)稳定性验证:建议评估在2~8 ℃保存条件下1周内(0、3和7 d)和-70~-20 ℃条件下1年内(0、3、6和12个月)质控品的使用稳定性[55 ]。
3. 室内质控判断规则:判断质控结果在控需同时满足:(1)阴阳性一致;(2)阳性质控荧光模型一致;(3)阳性质控品的滴度在靶值上下一个滴度范围内。在上述在控条件外,采用CAD系统进行滴度报告的实验室,可采用室内质控品的系统特异性荧光强度单位(light intensity unit,LIU),进行半定量监测,促进质量改进[54 , 56 , 57 , 58 ]。以变异系数(coefficient of variation,CV)为25%作为要求[54 , 56 ],弱阳性质控品建议使用Westgard规则12CV(单个质控点LIU超过平均数±2CV为警告)和13CV(单个质控点LIU超过平均数±3CV为失控),有利于监测整个试验过程以及发现系统误差[54 , 56 , 57 ]。
(二)室间质评
开展HEp-2 IFA检测ANA项目的实验室必须参加卫生主管部门组织的室间质评,以评估实验室对该项目的检测能力(包括荧光模型和滴度),如果室间质评中未涉及的必报荧光模型,建议定期进行实验室间比对。
(三)数据总结
患者数据对于HEp-2 IFA检测ANA的临床质量控制具有一定意义,是质控品的质量控制的补充。实验室可计算过去12个月ANA阳性率的均值,确立CV值,有利于系统误差的发现(如:不同人员间实验操作、结果判读的差异或批号间偏移等)[57 ]。
三、试剂批号验证
新批号试剂启用前必须完成批号验证。
(一)确认新批号分裂期细胞的数量
可采用有助于分裂期细胞识别的荧光模型,如:着丝点型(AC-3),NuMA样型(AC-26)或着丝点F样型(AC-14),确认是否有足够的分裂期细胞(3~5个分裂期细胞/高倍镜视野)。
(二)细胞密度确认
如果细胞密度过高或过低,应向制造商要求更换试剂批号。
(三)留样再测
不同批号试剂,由于一些蛋白的表达或者靶抗原与血清中自身抗体的结合能力可能有所差别,造成观察到的荧光模型不同。因此试剂新批号验证应包括室内质控品,阴性样本以及弱阳性和阳性样本,总样本数≥10份,符合率≥80%。阳性样本荧光模型应至少包括:均质型(AC-1),核颗粒型(AC-4,5),核点型(AC-6,7),核膜型(AC-11,12)和胞浆颗粒型(AC-19,20)[12 , 28 ]。建议实验室可收集一批上述荧光模型样本,保存于-20 ℃,用于新旧试剂批号比对。
结果报告
一、荧光模型报告
(一)必报荧光模型
2021年第六届ICAP工作会议对ANA荧光模型命名分类树进行修订,建议目前临床可报告荧光模型共30种(AC-0~AC-29),为确保具有重要临床意义的荧光模型被准确识别,ICAP将荧光模型分为必报荧光模型和选报荧光模型。基于HEp-2 IFA检测ANA荧光模型的国际分类和命名原则[59 ],结合国内检测的临床实践[60 ],本共识建议必报荧光模型16种包括:阴性(AC-0)、均质型(AC-1)、致密细颗粒型(AC-2)、着丝点型(AC-3)、核颗粒型(AC-4,5)、核点型(AC-6,7)、核仁型(AC-8,9,10)、核膜型(AC-11,12)、核多形性型(AC-13,14)、胞浆线性/肌动蛋白型(AC-15)、胞浆纤维型(AC-16,17)、胞浆颗粒型(AC-19,20)、胞浆网状/线粒体样型(AC-21)、胞浆极性/高尔基体样型(AC-22)、胞浆棒环状型(AC-23)和有丝分裂期荧光模型(AC-24,25,26,27,28)。30种临床可报告荧光模型及16种推荐必报荧光模型见表2。

(二)荧光模型判读的注意点
(1)做终点稀释的实验室应分别报告不同荧光模型的滴度;(2)以HEp-2细胞为基质的间接免疫荧光法检测ANA,必须以该基质细胞所呈现的荧光模型进行判读和报告[5 ],其他细胞基质,如猴肝细胞所额外表现出的荧光模型,可增加在备注信息中;(3)未被定义的荧光模型,可直接描述该荧光模型特征。
有一定判读经验的实验室读片人员,在完成相关培训及考核后,可报告30种荧光模型,但对报告荧光模型的相关临床意义应做好临床沟通。
(三)荧光模型滴度
推荐采用终点稀释法检测ANA荧光模型的滴度,不推荐依据起始筛查稀释度下的荧光亮度直接报告滴度结果。如图2所示,不同荧光模型在起始稀释度的荧光强度,与终点滴度的对应关系不同,仅依靠肉眼进行简单的滴度估算,主观性强且准确性低。当实验室仅进行起始筛查稀释度下单孔检测ANA时,推荐按照显微镜下的荧光强度报告半定量结果(图3),或依据终点稀释度报告终点滴度。部分采用CAD系统的实验室,可在性能验证通过后依据不同荧光模型报告CAD估算的终点滴度。
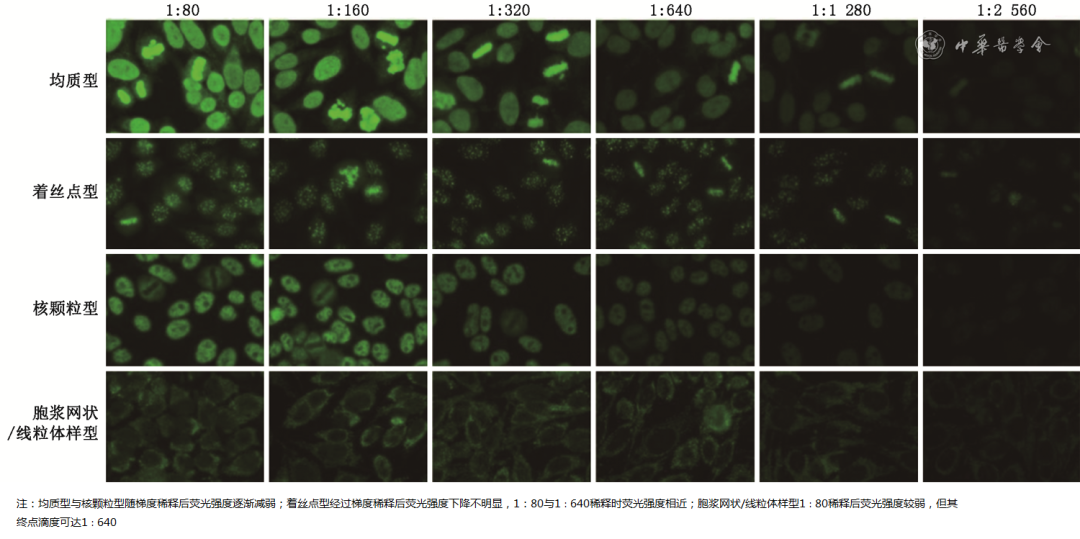
图2不同荧光模型梯度稀释终点滴度结果(间接免疫荧光染色×200)
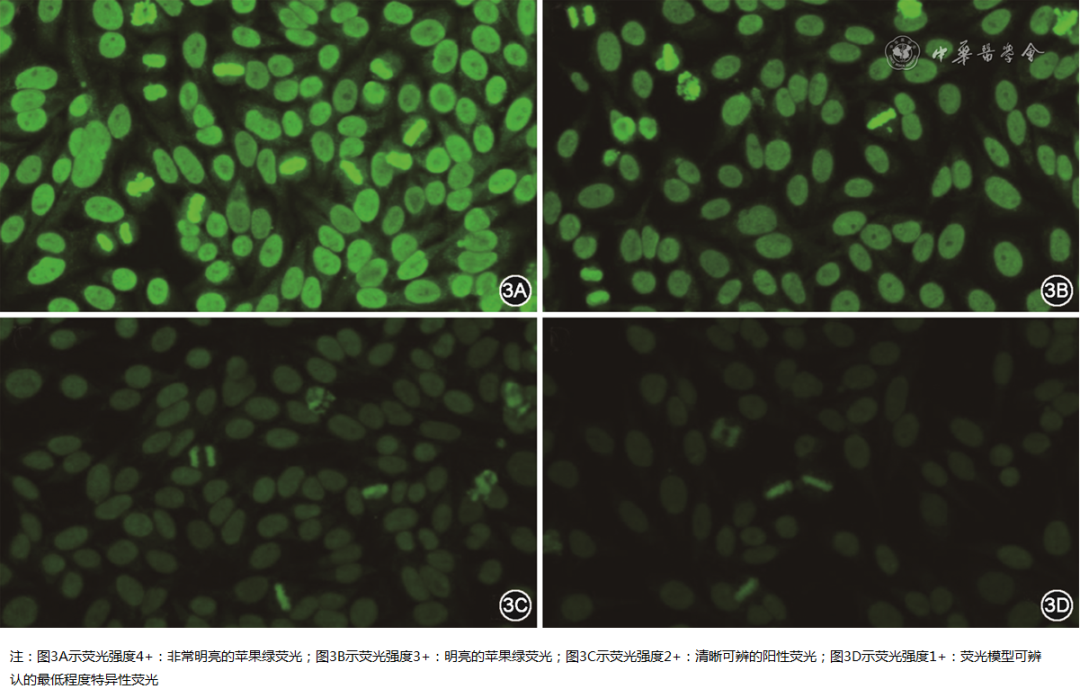
图3半定量荧光强度等级(间接免疫荧光染色×200)
二、滴度特异性似然比
似然比(likelihood ratios,LR)可同时反映诊断敏感性和特异性,即患者得出该筛查试验结果的概率与无病者得出这一概率的比值。例如:HEp-2 IFA检测ANA时,滴度1∶1 280检出了15% AARD患者和1%非AARD患者,那么1∶1 280滴度下LR为15(即15%除以1%)。LR等于1通常代表该滴度对于测试前和测试后概率之间没有差别,LR在5~10以及0.1~0.2代表测试前与测试后概率的临床意义中等程度差异,LR>10或<0.1则代表测试前与测试后概率的临床意义显著差异[61 ]。计算并报告LR可克服不同稀释体系的差异,提供对HEp-2 IFA结果的临床意义的估计,为临床医生提供重要信息[62 , 63 , 64 , 65 ]。
实验室可以通过文献查询或者回顾性分析AARD患者与非AARD人群滴度分布或CAD系统荧光强度,分别计算不同滴度或荧光强度下的LR,更能反映当地人群或特定人群(儿童或孕妇等)中滴度的临床意义。采用相同检测体系的实验室也可联合进行滴度特异性LR的多中心研究。
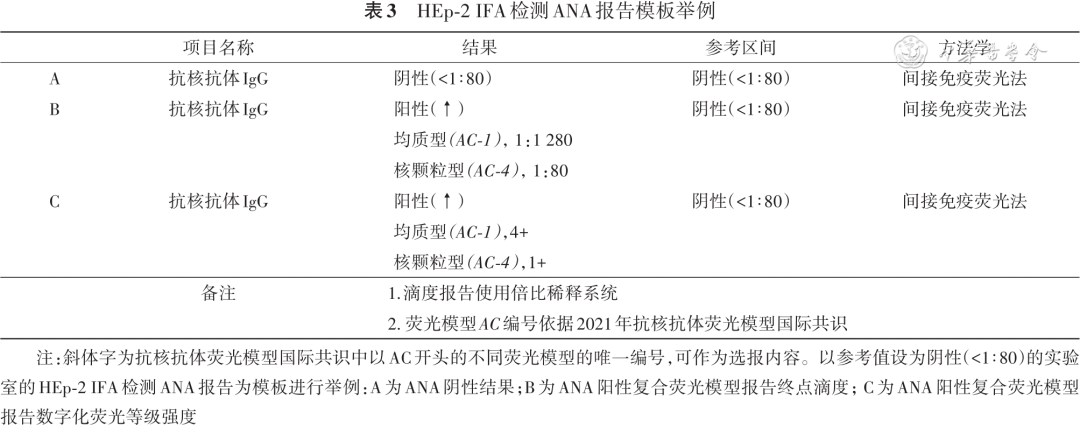
三、推荐的HEp-2 IFA检测ANA报告模板
报告的标准化对于临床医生解读报告以及检测结果的互认是十分必要的。表3列举了以1∶80作为筛查滴度的实验室HEp-2 IFA检测ANA报告模板,除了常规报告中所必需的患者基本信息、申请项目、申请科室及申请医生、诊断信息和样本类型,还需包括以下内容。
(一)检测方法学
检测ANA的方法学除了间接免疫荧光法外,还有化学发光法、酶联免疫吸附法等。不同检测方法敏感度和特异度不同,因此应在报告中明确检测方法学。
(二)复合荧光模型的报告
报告顺序按照细胞核荧光模型、细胞浆荧光模型和细胞有丝分裂期荧光模型。相同优先级荧光模型,按照滴度或与AARD临床相关性进行报告[5 , 60 ]。如:相同滴度情况下,致密细颗粒型与核点型混合模型,建议以核点型优先。
(三)滴度信息
HEp-2 IFA检测ANA为定性检测,因此报告需注明定性结果(阴性/阳性)。阳性报告还应有滴度信息。(1)荧光模型的终点滴度或荧光强度:做终点稀释法的实验室可报告不同荧光模型的终点滴度;仅做单孔稀释的实验室建议报告数字化荧光等级提示,如:“1+”~“4+”;或采用CAD系统报告主要荧光模型的估算滴度;(2)起始筛查稀释度:不同实验室采用的起始筛查滴度可能不同,应在报告中明确;同一实验室若对儿童和成人采用不同起始稀释度,也应在报告中注明。
(四)临床提示信息
报告单的备注部分是实验室与临床医生沟通的重要方式之一,如有相关复查信息,或必要的临床建议,可进一步说明。
结语
间接免疫荧光法作为抗核抗体检测的参考方法,在自身免疫病患者疾病诊断和评估中发挥重要作用。HEp-2 IFA检测ANA的影响因素较多,从实验室角度出发的专家共识对其规范化开展、标准化检测以及精细化报告具有重要意义,最终为临床提供可靠的ANA检测报告,并进一步精细化研究不同荧光模型在我国人群的临床意义提供依据。
主审专家:仲人前(原海军军医大学第二附属医院)
执笔人:郑冰(上海交通大学医学院附属仁济医院检验科),黄卓春(四川大学华西医院实验医学科),宁明哲(南京大学医学院附属鼓楼医院检验科)
专家组成员(按姓氏汉语拼音排序):蔡军(河南省人民医院检验科),车媛媛(吉林大学第一医院检验科),范列英(同济大学附属东方医院检验科),冯忠军(河北医科大学第三医院检验科),符克英(海南省人民医院检验科),耿燕(西安交通大学附属第二医院检验科),胡尧(复旦大学附属华山医院医学检验科),贾汝琳(北京大学人民医院风湿免疫科),金卫东(浙江省人民医院检验科),黎村艳(湖南省人民医院/湖南师范大学附属第一医院检验科),李立明(赣南医学院第一附属医院检验科),李敏(上海交通大学医学院附属仁济医院检验科),李一荣(武汉大学中南医院检验科),李永哲(中国医学科学院北京协和医院检验科),李志艳(北京大学第一医院检验科),刘义庆(山东第一医科大学附属省立医院临床医学检验部),刘治娟(西藏自治区人民医院检验科),罗静(山西医科大学第二医院风湿免疫科),牛倩(四川大学华西医院实验医学科),彭清林(中日友好医院风湿免疫科),彭奕冰(上海交通大学医学院附属瑞金医院检验科),宋超(浙江省临床检验中心),邰文琳(昆明医科大学第二附属医院检验科),唐志琴(天津市第一中心医院检验科),陶月(南京大学医学院附属鼓楼医院检验科),滕凤猛(江苏省中医院检验科),王健(广西医科大学第一附属医院检验科),王静(内蒙古医科大学附属医院风湿免疫科),王琰(青海红十字医院检验科),王艳萍(重庆医科大学附属第一医院检验科),吴丽娜(中国医科大学附属盛京医院检验科),徐菲莉(新疆医科大学附属中医医院检验科),尤崇革(兰州大学第二医院检验医学中心),余芳(贵州医科大学附属医院临床检验中心),曾智杰(中山大学附属第一医院检验科),张宏(中国科学技术大学附属第一医院检验科),张蜀澜(中国医学科学院北京协和医院风湿免疫科),张晓琍(福建医科大学附属协和医院检验科),张玉蓉(宁夏医科大学总医院医学实验中心),周海舟(哈尔滨医科大学附属第一医院检验科),朱宇清(上海市临床检验中心)
参考文献(略)
